本文来源于微信公众: 研学Biotech 作者: GENTOP
传统ADC已证明其价值,但也暴露了局限性:治疗窗口窄、靶向毒性、耐药性、异质性高等。新一代技术旨在通过以下核心特征解决这些问题:
1.高度均一性:从DAR随机分布的“混合物”变为DAR精确、位点特异的“单一产品”。
2.精准递送:利用更稳定的连接子和新型载荷,确保药物在正确时间、正确地点释放。
3.更强效力与新型作用机制:使用更强效的载荷或非细胞毒性药物,以克服耐药并扩大治疗领域。
4.增强肿瘤穿透与选择性:通过新型抗体格式或前药策略,提高对实体瘤的疗效并减少对正常组织的毒性。
新一代ADC技术正在从一门“艺术”转变为一门高度可控的“科学”。其发展趋势是:
- 设计理性化:基于对靶点生物学、载荷药理和连接子化学的深刻理解进行理性设计。
- 技术平台化:形成模块化的抗体、连接子、载荷平台,可快速组合成候选药物。
- 应用多元化:从晚期肿瘤治疗向更前线治疗、辅助治疗乃至非肿瘤领域拓展。
- 联合治疗常规化:与免疫检查点抑制剂、靶向药等联合使用,成为肿瘤综合治疗的基石方案。
最终目标是通过持续的技术迭代,开发出疗效更高、毒性更低、应用更广的“智能”靶向药物,为更多患者带来希望。
今天为大家解读一篇关于新一代ADC药物技术的文章。以期为大家在ADC药物研发设计方面提供参考。

1 引言
抗体偶联药物通过抗体的靶向特异性,选择性地将具有药理活性的小分子递送至病变细胞或组织,从而最大限度地减少对健康组织的不良影响。这种生物制品与小分子化合物的结合为疾病治疗提供了新的机遇,同时也给药物研发带来了独特的挑战。第一代ADC在临床上验证了有效性,同时也揭示了一些局限性,并推动了该技术的进一步改进。传统ADC主要使用auristatin或maytansinoid类微管蛋白抑制剂,通过表面赖氨酸或链间半胱氨酸随机连接至抗体。较新进入临床的ADC则包含了用于定点偶联的工程化半胱氨酸和新型DNA损伤剂。本文概述了已在临床前研究中展现出前景并有望转化进入临床的关键技术创新。这些创新包括更多的细胞毒性药物种类,如Tubulysins、对称和不对称苯并二氮杂䓬二聚体、蒽环类药物以及鹅膏毒肽。日益丰富的连接子可实现二硫键重桥连,以及高疏水性药物或高DAR值下的药物偶联。通过引入半胱氨酸、扩展遗传密码、聚糖工程和化学酶法,已开发出多种稳健的定点偶联方案。最后,探索递送调节免疫激活和脂质稳态的药物,用于癌症以外的适应症。总之,这些进展为增强和扩大ADC作为新型靶向疗法,治疗癌症及其他未满足医疗需求高的疾病的治疗带来了巨大希望。
下文将总结药物-连接子设计、抗体工程以及ADC在癌症领域外的潜在应用方面的最新进展。表1提供了ADC代表性技术和构建的概要。
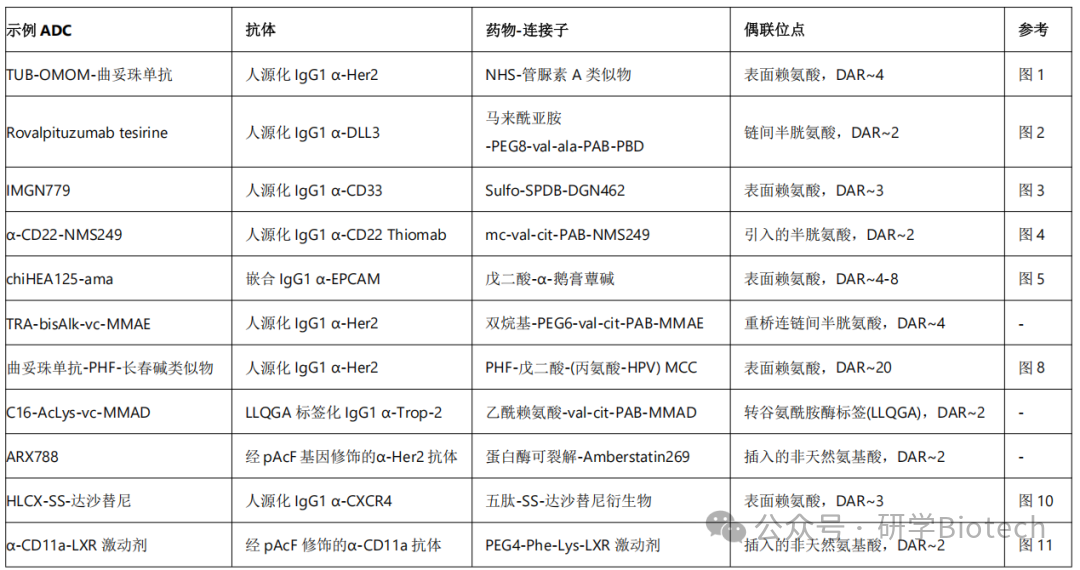
表1 ADC技术与示例
2 新型细胞毒素有效载荷和连接子
2.1 微管蛋白抑制剂
基于auristatin和maytansinoid的ADC药物的临床成功,促进了对其他靶向微管蛋白有效载荷的评估。每种新有效载荷都有望在效力、旁观者效应、耐药谱、副作用谱、偶联难易度等方面提供一套互补的特性。其他有效载荷包括新型auristatins、dolastatins和maytansines,以及新型化学结构类型,如隐藻素和Tubulysins。下文将以Tubulysins为例进行说明。
Tubulysins最初从粘细菌中分离,是一类独特的肽类化合物,能强效抑制微管蛋白聚合。许多不同的Tubulysins已从天然来源分离或通过合成获得。Tubulysin D(图1)是该类化合物中已知效力最强的成员之一,在多种癌细胞系中表现出皮摩尔级的细胞毒性活性。作为ADC有效载荷,一些Tubulysins既能对多药耐药肿瘤发挥强效活性,又能杀死旁观者细胞。天然Tubulysins的N,O-缩醛部分化学性质不稳定,促使人们寻找能保留高细胞毒性活性的更稳定的衍生物。Cohen等人成功制备了曲妥珠单抗与稳定的Tubulysin类似物Tub-OH和Tub-OMOM的偶联物。通过不可裂解的N-羟基琥珀酰亚胺连接子,将¹³¹I标记和未标记的Tubulysins与表面赖氨酸偶联。
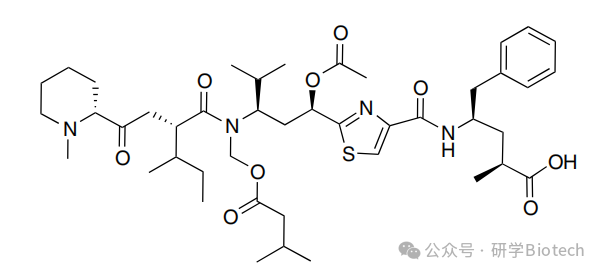
图1 tubulysin D的结构
2.2 苯并二氮杂䓬二聚体
吡咯并苯并二氮杂䓬二聚体是一类强效、序列选择性的DNA小沟结合剂,以SJG-136为例,图2所示。SJG-136也称为SG2000,在支持性护理下,以最大耐受剂量30µg/m²/天,连续给药3天(21天为一个周期),在晚期恶性肿瘤中显示出初步的抗肿瘤活性迹象。通常,每个PBD单体在N10-C11位置含有一个亲电亚胺基团,可与鸟嘌呤碱基的C2-NH₂基团共价连接。PBD能在低皮摩尔浓度下诱导DNA链间交联和细胞毒性。
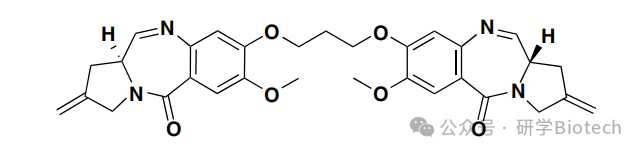
图2 SJG-136/SG2000结构示意图(一种吡咯并苯二氮䓬二聚体)
吲哚啉并苯并二氮杂䓬二聚体是一个小沟结合剂家族,具有亲电亚胺基团。与PBD类似,游离的IGN在体外和体内均具有强效细胞毒性。含有对称双亚胺IGN的ADC如预期般诱导了共价DNA加合物和交联;然而,小鼠治疗导致了延迟的毒性和死亡。相比之下,含有单亚胺的IGN仅诱导DNA烷基化。与基于IGN二聚体的ADC相比,含有可裂解二硫键连接子的单亚胺IGN抗CD33 ADC在体内表现出高效力,且早期和晚期毒性降低,图3所示。在DAR约为3时,该ADC显示出对急性髓系白血病异种移植瘤的最小有效剂量为0.6 mg/kg,在小鼠中的最大耐受剂量为40 mg/kg。
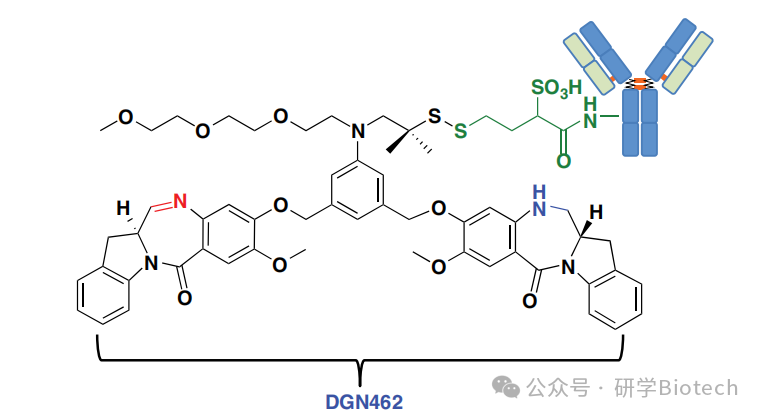
图3 IMGN-779结构示意图,一种含有吲哚并苯二氮䓬二聚体DGN462的抗CD33 ADC
2.3 蒽环类药物
这些DNA嵌入剂是应用最广泛的抗癌药物类别之一。蒽环类药物被认为通过阻断拓扑异构酶II的活性并诱导DNA双链断裂的形成来阻止DNA复制和转录。基于阿霉素的ADC是最早进行临床评估的ADC之一。然而,该化合物中等程度的效力、潜在的不可逆心脏毒性以及相关的耐药机制阻碍了其更广泛的开发。新型蒽环类药物如奈莫柔比星及其主要代谢物PNU-159682(图4)有潜力克服这些限制。与阿霉素相比,PNU-159682对一组癌细胞系的效力提高了1000倍,并且对具有多药耐药表型的细胞保留了活性。这些特性与PNU-159682对DNA异常紧密和稳定的结合有关。
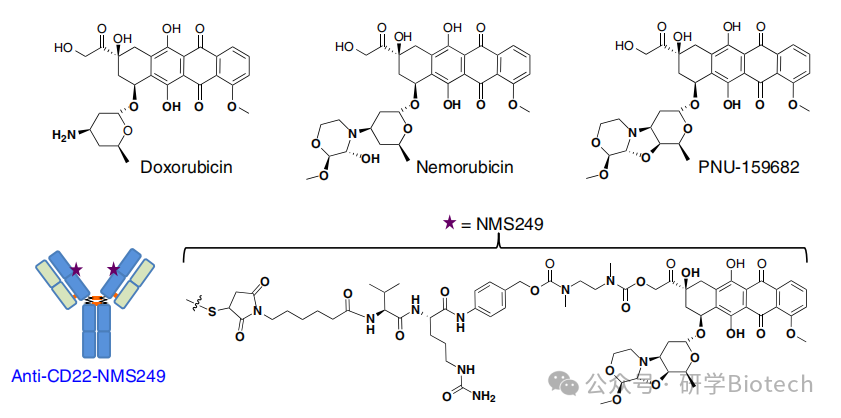
图4 强效蒽环类化合物与抗CD22-NMS249的结构
Genentech团队通过mc-vc-PAB和二乙胺连接子将PNU-159682与抗CD22单抗偶联得到CD22-NMS249(图4)。抗CD22抗体是pinatuzumab的THIOMAB版本。抗CD22-NMS249在体外对亲本非霍奇金淋巴瘤细胞系的效力比pinatuzumab vedotin强2至20倍。此外,单次1或2 mg/kg剂量的抗CD22-NMS249对四种NHL异种移植瘤均显示出体内疗效。与pinatuzumab vedotin不同,抗CD22-NMS249克服了由p-糖蛋白过表达或在pinatuzumab vedotin存在下连续传代诱导的耐药性,这与游离奈莫柔比星及其主要代谢物不是p-糖蛋白底物的发现一致。
2.4 鹅膏毒肽
这些化合物构成了一个双环八肽毒素家族。研究最深入的成员是α-鹅膏蕈碱(图5),由毒鹅膏菌产生。α-鹅膏蕈碱是真核生物RNA聚合酶II的强效特异性抑制剂。转录停滞会关闭翻译和正常代谢过程,引发增殖和静止肿瘤细胞的死亡。这种细胞毒性机制与当前临床阶段ADC的机制互补。
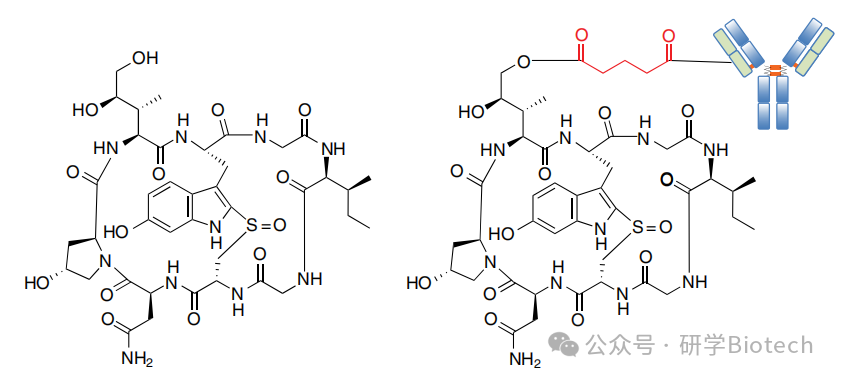
图5 α -鹅膏蕈肽与chiHEA125-ama ADC的结构
在一项研究中,α-鹅膏蕈碱通过戊二酸连接子与抗EpCAM单抗chiHEA125的表面赖氨酸偶联。在DAR为4-8时,该ADC对Colo205和MCF-7肿瘤细胞表现出个位数皮摩尔的效力,相对于非结合ADC具有惊人的100,000倍选择性。在BxPc-3胰腺癌异种移植模型中测试时,单次2 mg/kg剂量后观察到部分肿瘤消退,间隔一周给予两次4 mg/kg剂量后观察到完全消退。治疗通常耐受性良好,然而,单次6和12 mg/kg剂量与明显的肝毒性相关。这种副作用与意外鹅膏毒肽中毒后观察到的肝毒性一致。尽管α-鹅膏蕈碱的膜渗透性差,但它可通过肝脏特异性转运蛋白主动摄取。这个问题是鹅膏毒肽ADC项目的重要考量。
2.5 二硫键重桥连
目前处于临床测试的ADC中,大多数涉及通过抗体的链间半胱氨酸残基进行偶联。这种方法的局限性包括:对于DAR约4时形成异质混合物,对于DAR约2时存在大量未偶联的单抗,对于DAR约8时(使用传统药物-连接子)药代动力学和疗效欠佳,丢失了通常稳定重-轻和重-重链相互作用的共价键,以及传统马来酰亚胺连接子的血浆不稳定性。原则上,这些限制可以通过"重桥连"连接子克服,该连接子与原始二硫键的配对半胱氨酸形成共价键,这一策略最初是为蛋白质定点聚乙二醇化开发的。该方法涉及部分或完全还原链间二硫键,然后与药物-连接子反应,后者共价"桥接"来自原始二硫键的半胱氨酸硫原子(图6)。在大多数情况下,一个2或3碳的间隔基连接硫原子。这种方法的特点是,提供了定点特异性,而无需对抗体进行任何工程改造。
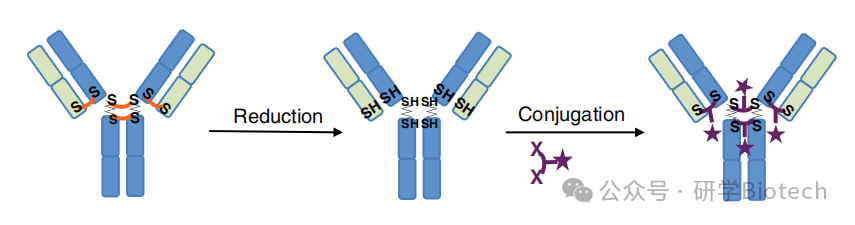
图6 二硫键重桥接策略示意图。星号表示通过连接子偶联的化学载荷,该连接子通过短(2或3碳)化学间隔基重新连接先前配对的半胱氨酸
关于这种策略的例子还有很多,例如图7所示。据报道,所得硫醚键在血浆中比使用马来酰亚胺化学形成的键更稳定,后者容易发生逆迈克尔反应。原则上,通过对完全还原的抗体进行重桥连,可以获得均质DAR=4的ADC,或使用Fabs或其他抗体支架获得均质DAR=1的偶联物。实际上,在某些情况下,链的完全重桥连仍然是一个挑战,部分原因是铰链区半胱氨酸之间形成链内桥,这些半胱氨酸仅由两个氨基酸隔开。使用更长的碳间隔基可能会加剧这个问题。
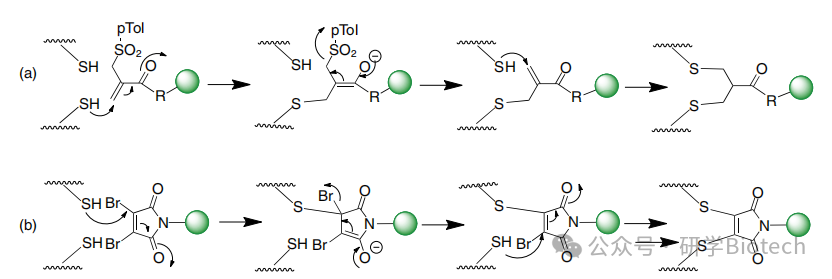
图7 基于单砜(a)或3,4-二溴马来酰亚胺(b)双烷基化反应的二硫键重桥接方法
2.6 Fleximer™聚合物连接子
多项研究已证明在使用传统药物-连接子时,具有确定且低DAR值(通常为2)的均质ADC的优势。然而,Mersana采取了不同的策略,使用亲水性聚合物连接子生成具有高且异质DAR值的ADC。这种可生物降解的聚合物连接子最初是为了提供一种喜树碱前药衍生物,以克服该分子的药代动力学限制。Mersana后来将这项技术用作克服某些ADC有效载荷疏水性限制的手段,并生成了DAR值在15至20范围内的ADC(图8)。这些ADC在生理pH下相对稳定,并在内化进入酸性内体后释放有效载荷。使用Fleximer™在DAR约20时将长春花碱类似物连接到曲妥珠单抗,未观察到Her2结合亲和力有明显损失。基于曲妥珠单抗的ADC单次20 mg/kg剂量导致BT-474人乳腺癌异种移植瘤完全消退,治疗后60天,100%的动物检测不到肿瘤。相比之下,使用非结合ADC或未偶联曲妥珠单抗与药物-连接子的混合物治疗后,仅观察到轻微的肿瘤生长延迟,没有肿瘤消退。
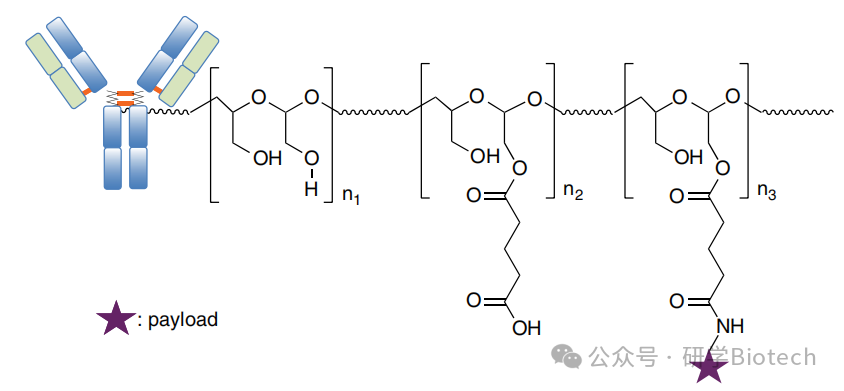
图8 基于Fleximer聚合物技术的ADC基本结构
3 抗体改造
药物-连接子化学的创新与改造抗体的新方法相辅相成,以促进药物偶联。其中一个主要目的是生成具有确定DAR值的均质ADC。如图9所示,已经开发了几种策略来使用工程化抗体或酶介导的偶联制备定点特异性ADC。定点特异性ADC表现出改善的性能,主要是因为没有快速清除的高DAR物种,并且与通过半胱氨酸硫醇进行马来酰亚胺偶联相比,消除了逆迈克尔反应。
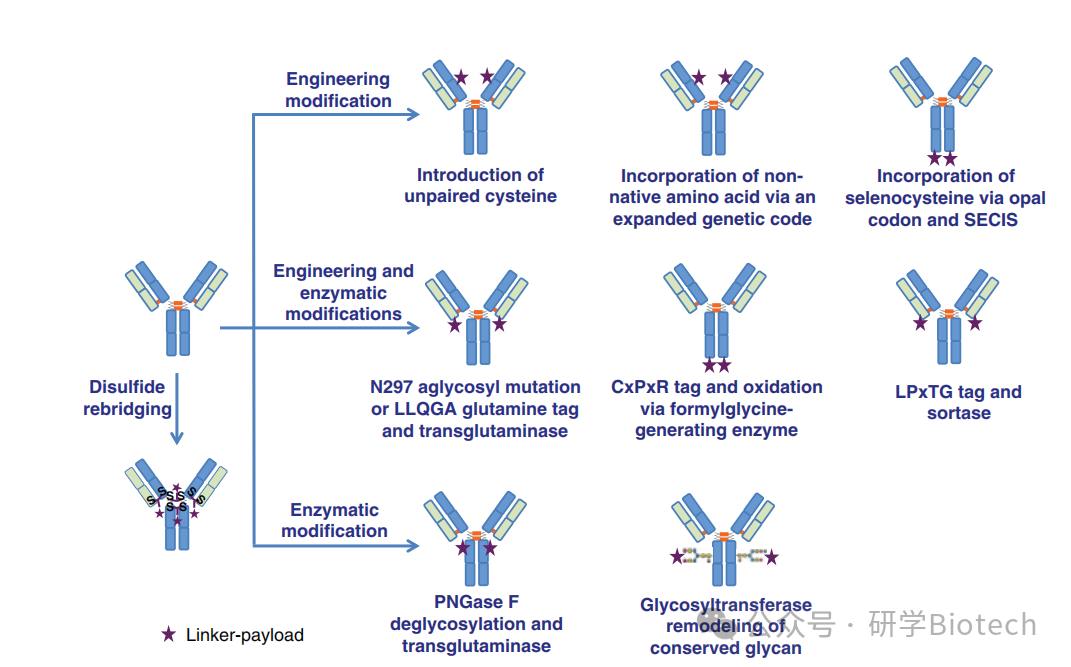
图9 位点特异性ADC的制备方法示意图( SECIS =硒代半胱氨酸插入序列)
3.1 工程化半胱氨酸
人IgG的恒定区不存在不成对半胱氨酸,可变区也很少出现。因此,引入的半胱氨酸可以提供可利用的化学手柄。然而,不成对半胱氨酸可能导致表达/分泌减少、蛋白质聚集、二硫键混乱以及其他生产挑战。这些因素限制了引入半胱氨酸的优选位点数量,并促使人们经验性地寻找对特定应用最优的位点。
Junutula等人率先利用工程化半胱氨酸制备定点特异性ADC,即THIOMABs。这项工作利用了之前开发的噬菌体展示方法,来识别将活性半胱氨酸插入抗体Fab片段的位点。在全长人IgG1背景下评估了轻链上的8个位点和重链上的4个位点,发现重链CH1结构域中的A114C位点表现出良好的表达和偶联效率。即使在这个优选位点,发现引入的半胱氨酸在表达和纯化后与谷胱甘肽、游离半胱氨酸等其他含硫醇化合物甚至抗体轻链形成二硫键连接。对于偶联,将所有溶剂可及的(即引入的和链间的)二硫键还原,然后使用硫酸铜或脱氢抗坏血酸重新氧化链间二硫键。
与传统ADC相比,TDC在体外表现出相当的结合亲和力,并且在OVCAR-3等卵巢癌模型中表现出相似或更好的临床前疗效。单次6 mg/kg剂量的TDC在每个模型中都能导致已建立肿瘤的消退或长期控制。重要的是,TDC在大鼠中表现出更优的药代动力学,在大鼠和食蟹猴中的耐受性提高了三到四倍。
工程化半胱氨酸显著提高了与马来酰亚胺形成的硫醚键的稳定性。研究发现,带正电荷的环境促进了琥珀酰亚胺环的水解,开环最大限度地减少了因逆迈克尔反应导致的药物损失可能性。这项研究还检验了将半胱氨酸引入抗体Fc区的影响。除了MUC16,针对Her2、STEAP1和CD22的抗体,在使用MMAE、DM1和蒽环类有效载荷时,TDC也显示出优于传统ADC的优势。
3.2 酶辅助偶联
定点偶联的另一种策略是利用酶对其底物的特异性。该方法利用了甲酰甘氨酸生成酶、转谷氨酰胺酶、分选酶和葡萄糖基转移酶来修饰抗体的多肽或聚糖结构。在某些情况下,首先通过引入内部或末端寡肽标签来修饰抗体序列以促进催化。引入标签的潜在免疫原性是一个理论上的关注点。
3.2.1 微生物转谷氨酰胺酶
转谷氨酰胺酶(TG)催化伯胺与谷氨酰胺的γ-羧酰胺之间形成异肽键。TG几乎存在于所有生物体中。来自放线菌的微生物TG广泛用于食品工业,并曾用于生物素、聚乙二醇和放射性金属螯合剂修饰蛋白质。在这些早期研究中,对全长鼠源单抗和肽标签化的单链Fv抗体进行了TG标记。研究证实,这种微生物TG对基于胺的底物具有混杂性,但对蛋白质谷氨酰胺的识别更具限制性。
Schibli及其同事研究了将荧光团与天然和去糖基化形式的嵌合IgG1抗L1-CAM抗体chCE7的偶联。尽管最大取代率为每个mAb一个荧光团,但与去糖基化mAb的偶联效率明显更高。该小组的后续研究表明,酶法去糖基化的chCE7优先在Q295位点被修饰,而N297Q去糖基化突变体优先在Q295和Q297位点被修饰。推测对这些谷氨酰胺的特异性源于它们位于人IgG1的柔性区域。
Rinat在人IgG1的90个表面可及位点插入了谷氨酰胺标签。12个位点适合偶联,研究重点分别是在重链或轻链C末端添加LLQGA或GGLLQGA。使用抗M1S1抗体和通过可裂解AcLys-vc-PAB连接子连接的MMAD,制备了定点特异性和传统ADC。通过TG介导的重链或轻链标签偶联,在体外观察到等效的效力,以及对小鼠BxPC3胰腺癌异种移植瘤的等效疗效;然而,轻链偶联在大鼠血浆中表现出更优的药代动力学和稳定性。大鼠血浆中的不稳定性归因于缬氨酸-瓜氨酸二肽的裂解,而不是TG异肽键的降解。相反,对于使用C6-vc-PAB连接子和新型auristatin抑制剂Aur-0101制备的ADC,在小鼠血浆中观察到比大鼠血浆中更大的不稳定性;然而,在猴和人血浆中观察到了良好的稳定性。使用TG制备了高度均质的ADC;然而,为了消除对天然谷氨酰胺的非靶向偶联,通过将Q295突变为天冬酰胺获得了最佳结果。
3.2.2 甲酰甘氨酸生成酶
FGE是一个广泛分布的酶家族,催化I型硫酸酯酶活性位点内半胱氨酸在翻译过程中氧化为甲酰甘氨酸。半胱氨酸侧链从─CH2SH转化为─CH2CH═O对于细胞硫酸酯酶的活性至关重要。醛基提供了相对于20种标准氨基酸的正交官能团。哺乳动物和细菌的FGE都识别一个五聚体CxPxR共有序列,该序列可以插入异源蛋白质中。
FGE标签在插入抗体重链和轻链的内部和羧基末端时都表现出耐受性。一种优选方法是在重链羧基末端用SLCTPSRGS(FGE共有序列加粗)替换赖氨酸。为了补充内源仓鼠FGE,抗体重链和轻链在同时过表达人FGE的中国仓鼠卵巢细胞中表达。含甲酰甘氨酸的抗体适合进行经典醛基反应以形成肟和腙偶联物。为了提高血浆稳定性,已使用Pictet-Spengler、HIPS和trapped-Knoevenagel连接来生成抗体和药物-连接子之间高度稳定的C─C键。
曲妥珠单抗在重链羧基末端进行了醛标记,并与HIPS-谷氨酸-PEG2美登素药物-连接子偶联。通过疏水相互作用色谱去除未偶联的单抗后,获得了DAR为2的均质ADC。与使用传统赖氨酸化学制备的曲妥珠单抗-DM1(DAR=3.4)相比,该定点特异性ADC在体外细胞杀伤活性和对NCI-N87异种移植瘤的单次剂量体内疗效方面表现出相似或更好的效果。在小鼠中,定点特异性ADC的血清半衰期和暴露量比传统ADC高30-40%,但低于未修饰的曲妥珠单抗。此外,在大鼠中以等效单次剂量给药时,定点特异性ADC表现出比传统ADC更低的毒性。正如预期,两种ADC的毒性性质相似。
3.2.3 葡萄糖基转移酶和其他聚糖工程
Asn-297是人IgG恒定区内N-连接糖基化的唯一规范位点。由于其远离抗原结合位点且对抗体药代动力学影响最小,聚糖是具有吸引力的偶联位点。用高碘酸盐氧化天然聚糖产生活性醛基是常见的方法;然而,反应条件可能导致异质混合物、蛋氨酸残基氧化以及结合活性丧失。
SynAffix描述了一种三步程序,用于制备具有精确定义DAR值的聚糖偶联ADC。在一个例子中,首先用内切糖苷酶Endo S处理天然抗体,该酶切割N-连接聚糖,留下单个GlcNAc,如果存在则保留岩藻糖残基。接下来,使用牛β(1-4)-半乳糖基转移酶的Y289L突变体将含叠氮的糖转移到抗体上。然后通过点击化学连接药物-连接子,得到DAR=2的ADC。
Zhou等人描述了一种替代程序,即首先在UDP-半乳糖和CMP-唾液酸存在下,使用β(1-4)-半乳糖基转移酶和α(2-6)-唾液酸转移酶对抗体进行唾液酸化。温和的高碘酸盐氧化后,含醛基的抗体通过肟化学进行偶联。使用这种方法,曲妥珠单抗与氨基氧基-Cys-mc-vc-PAB-MMAE药物-连接子偶联,生成DAR约2的ADC,该ADC包含具有0、1、2及更高DAR值的混合物。
Okeley等人描述了一种非酶促的代谢方法来改造抗体。将目标抗体在有含硫醇岩藻糖类似物的培养基中表达,该类似物被整合到Asn-297聚糖中。游离硫醇使得能够使用标准马来酰亚胺化学与mc-vc-PAB-MMAE偶联证实偶联仅通过重链发生,得到的DAR约1.3。
3.3 非天然氨基酸和硒代半胱氨酸
另一种方法利用扩展的遗传密码,该密码包含相对于20种标准氨基酸具有正交反应性的非天然氨基酸。在这种方法中,通过工程化的tRNA和氨酰-tRNA合成酶对,将琥珀终止密码子重新定向以掺入非天然氨基酸。迄今为止,已有超过100种不同的非天然氨基酸被掺入蛋白质中。这种方法已被用于在大肠杆菌和哺乳动物细胞中体外表达的抗体。尽管琥珀终止密码子在哺乳动物细胞中广泛使用,但从稳定转染的CHO细胞中已能生产克/升级别的含非天然氨基酸的抗体。
曲妥珠单抗在重链A114位点用pAcF修饰,并通过含有三个乙二醇单元和非裂解连接子与MMAF偶联。使用传统马来酰亚胺化学通过链间半胱氨酸将野生型曲妥珠单抗与MMAF偶联作为对照。非天然氨基酸ADC和传统ADC的DAR分别为2.0和3.8。尽管DAR较低,非天然氨基酸ADC在体外表现出与传统ADC相当的细胞毒性,并且在NCI-N87和HCC-1954 Her2+异种移植模型中表现出相似或更好的疗效。此外,在死亡率、体重、血液学和血清化学方面,非天然氨基酸ADC显示出更好的耐受性。总体而言,与传统ADC相比,非天然氨基酸ADC的治疗窗口至少扩大了两倍,部分原因是其更优的血浆稳定性和药代动力学。
Sec通常被称为遗传密码中的"第21种氨基酸",自然存在于约25种人类蛋白质中,其中许多充当氧化还原酶。Sec在UGA终止密码子处的掺入是通过专用的tRNA、3' sec插入序列和SECIS结合蛋白的协调作用实现的。与半胱氨酸相比,Sec在弱酸性pH值下比硫醇盐对应物更具反应性的亲核试剂,从而能够选择性标记Sec。此外,与马来酰亚胺硫醚键相比,与Sec的砜偶联可以提供更好的血浆稳定性。
3.4 替代形式和掩蔽抗体
全长IgG具有血清半衰期长、肾或肝胆清除有限以及免疫原性低的优点。然而,与此形成鲜明对比的是,单链Fv、Fabs和其他较小的抗体支架仍然是临床阶段免疫毒素的主力,这些免疫毒素使用基于蛋白质的毒素作为细胞毒性部分。靶向CD22和CD25的免疫毒素在不同类型的白血病早期临床研究中显示出持久的反应。较小的支架可以改善通常受扩散限制的肿瘤渗透。抗体的扩散性被认为与大小成反比,因此单链可变区片段抗体相对于全长抗体具有约六倍的优势。此外,较小的结构可以为定点偶联提供更多机会。
这些构建体包括scFvs、双特异性抗体、Fabs和同源二聚体单链抗体。在每种情况下,都是通过表面暴露的半胱氨酸残基实现定点偶联。Perrino等人测试了一种名为SIP(F8)-SS-DM1的ADC,其中抗体成分是一个靶向纤连蛋白剪接变体的同源二聚体"小免疫蛋白"。该ADC是一种非内化ADC,设计用于结合肿瘤血管系统中的EDA,并在二硫键还原后在局部释放高浓度DM1。在F9畸胎癌小鼠模型中,7 mg/kg q3dx3方案的耐受性良好,并伴有肿瘤完全消退。
在大多数情况下,实体瘤ADC面临着靶点在正常组织上表达的挑战。正常组织的表达可能导致剂量限制性的靶上毒性,或通过增加ADC在肿瘤外的摄取和有效载荷释放而导致脱靶毒性。Probody™技术为缓解此问题提供了一种方法。在Probody中,抗体轻链被修饰成包含一个氨基末端掩蔽肽和蛋白酶可裂解的连接子。掩蔽肽旨在阻断正常组织中的抗原结合,但在肿瘤微环境中被蛋白酶释放。在小鼠EGFR表达异种移植瘤模型中,野生型和Probody形式的西妥昔单抗显示出相似的抗肿瘤疗效。然而,在非人灵长类动物中,Probody显示出更好的安全性和循环半衰期。
3.5 ADC在肿瘤学以外适应症的应用
除了Adcetris™在系统性红斑狼疮患者中的探索性II期研究外,所有临床阶段的ADC都携带用于癌症治疗的细胞毒性有效载荷。从概念上讲,细胞毒性ADC可以更广泛地开发用于肿瘤学以外的用途,之前在炎症、感染和眼部疾病中对免疫毒素的临床评估为此提供了先例。然而,更重要的是,抗体有潜力特异性递送其他类别的药物,同样的原理适用于将高活性化合物特异性递送至目标细胞,同时最大限度地减少对非靶细胞和组织的不良影响。为此,临床前研究评估了靶向CD163+巨噬细胞的地塞米松、靶向人类免疫缺陷病毒的CCR5拮抗剂、靶向CXCR4+造血细胞的多激酶抑制剂达沙替尼以及靶向CD11a+巨噬细胞的LXR激动剂。
达沙替尼(图10)通过抑制bcr-abl激酶,用于治疗费城染色体阳性白血病。达沙替尼还抑制其他几种激酶,包括介导T细胞信号传导的src家族成员lck和fyn。然而,作为游离药物的达沙替尼的副作用谱阻碍了其作为免疫抑制剂的评估。CXCR4是一种趋化因子受体,在多种造血和非造血细胞类型上表达。Wang等人使用基于二硫键和非裂解连接子(DAR约3)将达沙替尼与抗CXCR4和对照抗体偶联。测试了这些ADC在抗CD3/抗CD28抗体刺激下对原代人T细胞分泌TNF-α和IL-2的影响。可裂解的抗CXCR4 ADC在13-27 nM浓度下阻断50%的细胞因子分泌,而非结合ADC在200 nM时影响甚微。同样,未偶联的抗体和游离药物-连接子在300 nM浓度下均无活性。需要进一步研究以表征这些ADC在相关疾病模型中的安全性和有效性。
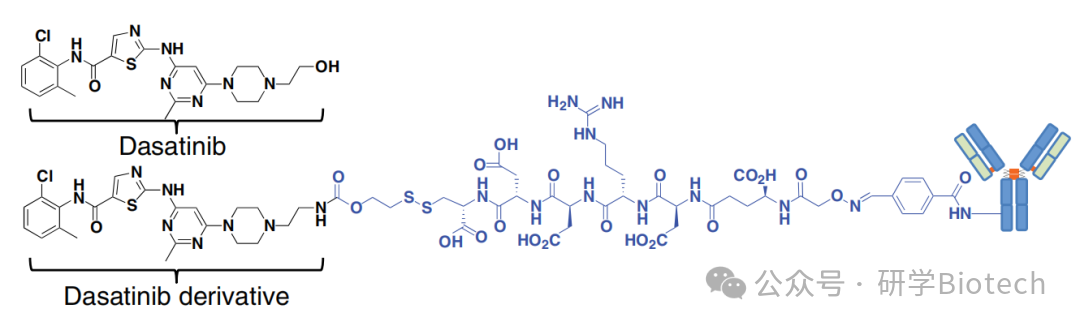
图10 达沙替尼及其可裂解的基于达沙替尼的抗CXCR4 ADC的结构(用于靶向免疫抑制)
基于LXR激动剂在促进巨噬细胞胆固醇流出方面的有益作用,人们一直在探索将其用于动脉粥样硬化治疗。然而,这些药物在肝脏中的不良靶向效应阻碍了其临床开发。为了赋予巨噬细胞特异性,Lim等人将一种强效LXR激动剂与抗CD11a抗体偶联。CD11a是整合素LFA-1的α亚基,在单核细胞、巨噬细胞和其他白细胞上广泛表达。如前所述,有效载荷通过定点特异性方式与已工程化含有非天然氨基酸pAcF的抗CD11a和对照抗体偶联。与非结合对照ADC不同,抗CD11a ADC(图11)特异性结合并被CD11a+ THP-1单核细胞(而非CD11a- HepG2肝癌细胞)内化。抗CD11a ADC在THP-1(而非HepG2)报告细胞系中诱导了LXR驱动的荧光素酶表达,而游离药物在两系中均有活性。
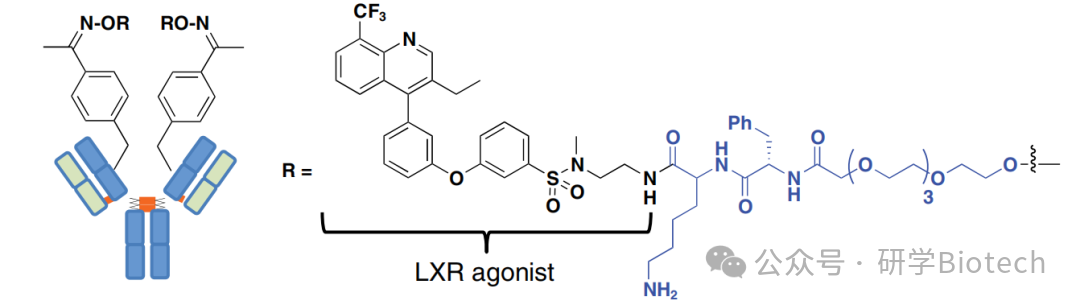
图11 抗CD11a- LXR 激动剂抗体偶联药物(ADC)结构示意图
4 结论
抗体工程和药物-连接子设计的单独和协同创新,已在临床前模型中显著提高了肿瘤靶向ADC的活性广度和治疗窗口。另外,未来的方向可能包括增强效力、选择性或内化的双特异性ADC、携带多类药物的ADC。此外,药物分子可以被设计为在细胞外发挥作用,以实现靶向特异性或调节受体活性。ADC技术的快速发展与优化,为改善多种人类疾病的治疗提供了新的方案。