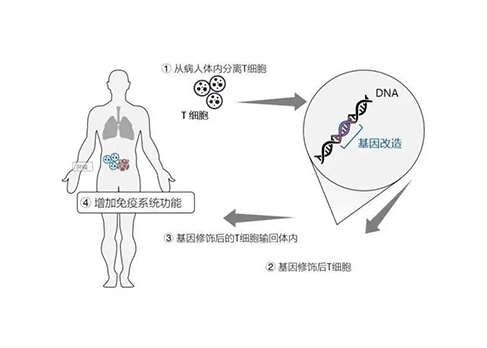
本文来源于微信公众: 医药学术 作者: 医药学术编辑部
CAR-T疗法在血液瘤里算是杀出了一片天。但一碰到实体瘤,就仿佛拳头打进了棉花里—使不上劲。为什么呢?主要有两大“拦路虎”:一是肿瘤细胞的异质性,二是肿瘤微环境这个“保护壳”。实体瘤就像个坚固的堡垒,外面有乱七八糟的血管和基质细胞挡着,CAR-T细胞很难钻进去;就算进去了,里面还充斥着各种抑制免疫的信号和细胞,活活把CAR-T细胞“耗死”或“管住”。今天我们就系统梳理这些障碍,并总结一下目前最有希望的破解之道。
克服抗原逃逸和异质性的策略
肿瘤细胞抗原丢失或者不统一怎么办?有以下几种应对策略:广撒网:设计能同时识别两个靶点的CAR-T细胞,或者让CAR-T细胞分泌“双特异性T细胞衔接器”(BiTE)的小抗体,把更多肿瘤细胞甚至 bystander T细胞都拉进来打架。车轮战:序贯输注两种不同靶点的CAR-T,这个打完那个上,不让肿瘤喘息。
这里提到一个有趣又麻烦的现象—“胞啃作用”。CAR-T细胞和肿瘤细胞近距离接触时,会互相撕扯膜蛋白。结果可能是:肿瘤细胞把CAR分子抢走了,把自己伪装成CAR-T细胞,导致真CAR-T细胞认不出它(抗原掩蔽);或者CAR-T细胞把肿瘤抗原抢走了,导致自己人打自己人( fratricide,兄弟相残)。应对策略包括优化CAR的敏感性、改造共刺激域等。抓弱点:对于靶点表达量很低的肿瘤细胞,可以通过优化CAR的结构来提升CAR-T细胞的识别灵敏度。
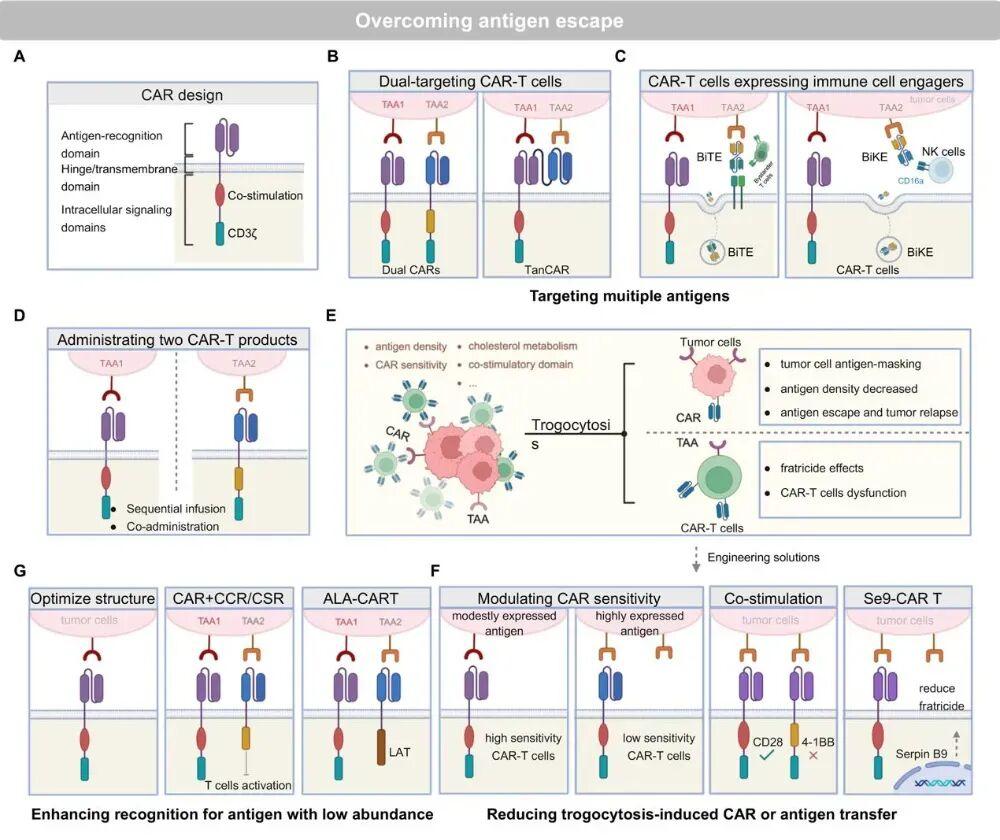
克服肿瘤细胞免疫逃逸的策略
阻断PD-1/PD-L1通路增强CAR-T
肿瘤利用PD-1/PD-L1这个“刹车”系统抑制CAR-T。我们的策略是直接改造这个系统:
信号转换:给CAR-T加上PD-1/CD28转换受体。当肿瘤的PD-L1来结合时,本来应该传递抑制信号,但这个改装受体却把它转换成激活信号CD28,相当于把敌人送来的“毒酒”换成了“兴奋剂”。
基因敲除/下调:直接用基因编辑技术敲除CAR-T细胞自身的PD-1基因,或者用RNA干扰技术降低其表达。
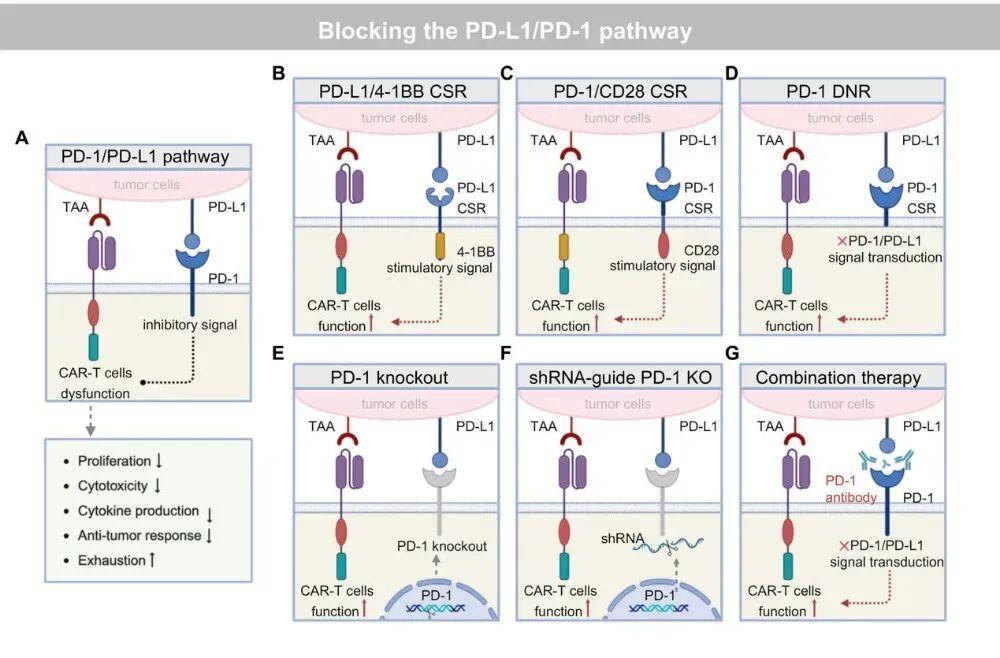
阻断PD-1/PD-L1通路以增强CAR-T疗效
靶向肿瘤微环境和增强CAR-T自身续航
改善交通与渗透:给CAR-T装上肿瘤微环境中高表达的趋化因子受体,就像给它一个准确的GPS导航,让它能沿着化学信号浓度梯度快速抵达肿瘤。拆除物理屏障:开发靶向CAFs或细胞外基质的CAR-T。或者让CAR-T分泌基质降解酶,像带着“破拆工具”一样溶解屏障。
中和抑制信号:重点对付TME中的“头号抑制因子”TGF-β。可以给CAR-T表达负性TGF-β受体,使TGF-β信号无效;或者更巧妙地设计一个反向细胞因子受体,把接收到的TGF-β抑制信号逆转成促进CAR-T增殖的IL-15信号。
优化CAR-T自身“续航”:优化共刺激域:经典的CD28域让CAR-T反应猛但容易耗竭;4-1BB域则利于长期存活和记忆形成。现在也在探索OX40、ICOS、CD27等新型共刺激域,或者将它们组合使用。巧用细胞因子:让CAR-T自产自销细胞因子,在肿瘤局部形成支持自身存活和扩增的微环境,避免全身给药的毒副作用。调控分化状态:通过过表达c-Jun、TCF1等转录因子,或敲除NR4A、TOX等耗竭相关因子,让CAR-T维持在更年轻、更有潜能的“干细胞样记忆T细胞”状态,而不是容易耗竭的终末效应状态。
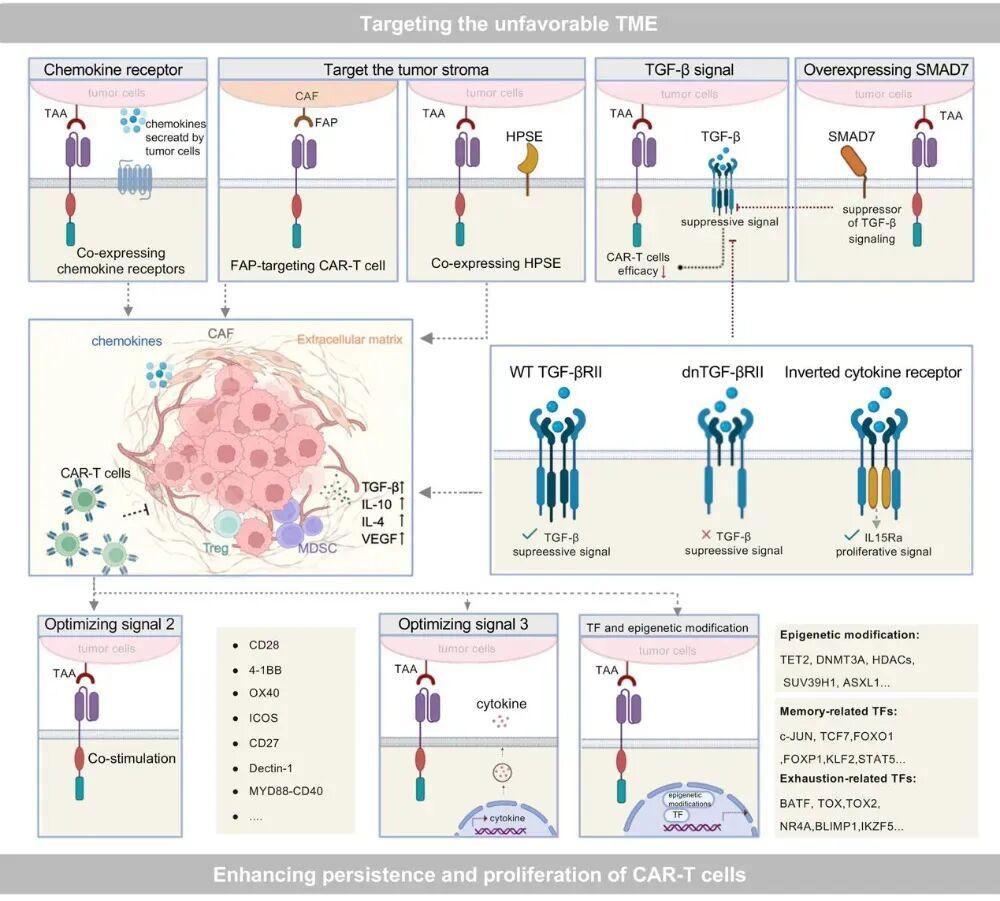
靶向TME并增强CAR-T持久性与增殖的策略
未来方向—联合治疗是王道
面对如此复杂的实体瘤,单靠某一项CAR-T技术改造很难成功,必须多策略联用,甚至与其他疗法强强联合。例如,联合免疫检查点抑制剂,解除全身性免疫抑制。联合放疗,放疗能破坏肿瘤结构、增加抗原释放、改善局部微环境,为CAR-T进入创造条件。联合溶瘤病毒,病毒能特异性感染并裂解肿瘤,同时改造TME,还能携带一些基因来辅助CAR-T。
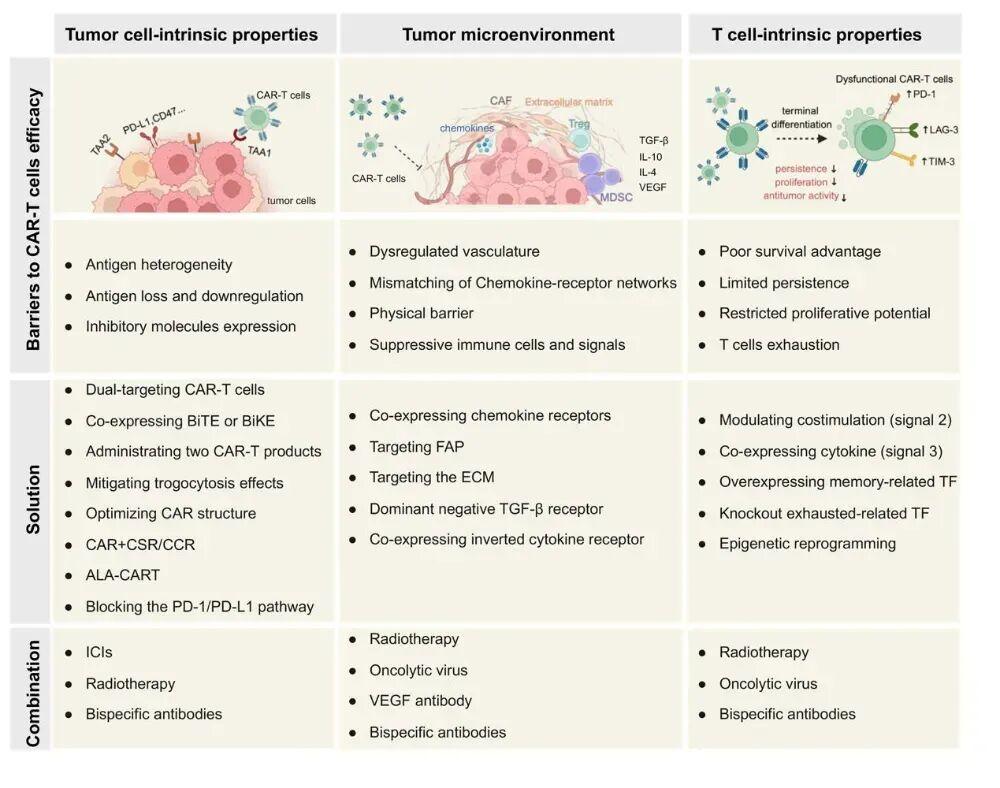
下一代CAR-T细胞设计与联合策略
对抗实体瘤,新一代CAR-T的设计思路不再是简单的“识别-杀伤”,而是进化为一个集多靶点、渗透导航、破除屏障、抵抗抑制、自我续航”于一体的综合性活体药物平台,并且需要与放疗、免疫药物等手段协同作战。