文章来源公众号:有理就听你的 作者:有理就听你的
阿斯利康的科学家指出:靶点验证的可信度及靶点结合生物标志物的应用,能显著提升化合物通过II期临床试验的概率。
辉瑞更是提出著名的“3 pillar范式”,明确表示,药物需满足以下三个条件,临床成功率可达80%以上:
·Pillar 1: 作用部位的暴露(Exposure at site of action):即药物能够到达目标部位(如病灶组织、细胞),并维持有效浓度。
·Pillar 2: 与药理靶点的结合(Target binding):即药物需与预设靶点(如受体、酶)特异性结合,触发药理效应。
·Pillar 3: 作用部位的药理活性表达(Pharmacological activity):即在目标部位,药物需达到足够浓度并激活下游通路,最终转化为治疗效应。
这里就要说了Target Engagement ,不仅是药物研发的“验证基石”,更是贯穿从靶点确认、临床前研究到临床试验的核心指标。其通过量化药物与靶点的结合特性,显著降低研发风险,提升成功率,并为个体化治疗提供科学依据。
本文为靶点结合检测(Target Engagement Assays)的选择提供了系统性框架,旨在解决临床前药物研发中的一个关键决策难点。该框架将成为科研人员在检测方法选择中实现理性、快速且无偏决策的关键资源,以加速药物研发进程。
对各类疾病生物学机制的深入理解,以及单蛋白制备、分离与分析技术的进步,基于靶点的药物研发(TBDD)近几十年来已成为制药行业的主导策略。在选定并验证与疾病相关的生物靶点后:
(1) TBDD的核心在于:设计特异性结合靶点蛋白的化合物以诱导治疗效应,理想状态下需实现最小脱靶效应且无副作用。为此,需开发能监测蛋白与配体间结合(即靶点结合)的检测方法,并量化相互作用强度,以迭代优化分子与靶点的结合能力,构建构效关系(SAR)。
(2) 此类检测为候选药物的作用机制(MoA)提供证据,而MoA已被证实与改善临床结局直接相关。
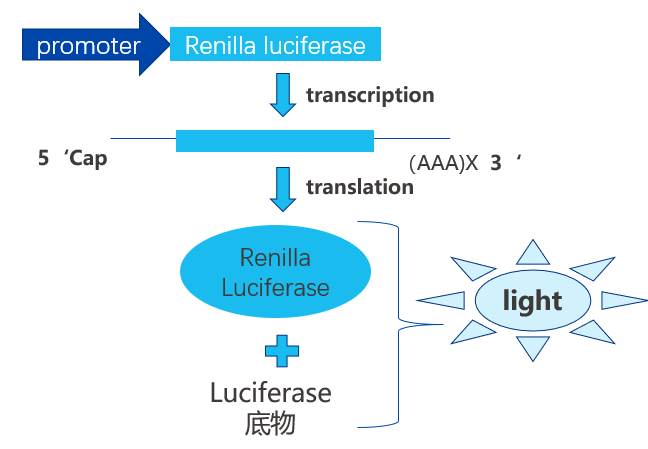
靶点结合(Target Engagement)可通过开发特定检测体系进行量化:向靶点蛋白中加入配体后,基于蛋白-配体相互作用程度生成可定量读数。由于配体结合通常引发物理性质变化,因此可直接使用多种生物物理技术测定靶点结合(本《视角》将详述这些技术)。此外,也可通过生化活性检测间接测量,例如监测酶促反应产物的浓度变化,或配体与受体结合后的下游效应。此类功能检测虽未纳入本文讨论范畴(因其对特定蛋白具有高度特异性),但在检测级联中仍至关重要——其不仅能验证药物是否结合靶点,还能确认是否产生预期药理效应。功能检测与直接结合监测结果的对比分析,可进一步证实观测效应确由药物-靶点相互作用引发,并揭示相互作用的效率。
现有大量靶点结合检测技术用于解析化合物与离体蛋白(通常为重组蛋白)的相互作用。尽管基于离体蛋白的检测对小分子迭代优化有重要作用,但其高度简化模型难以反映蛋白在体内的真实行为。鉴于人类蛋白质组的庞大规模和复杂性,建立药物分子在细胞内特异性结合预期靶点的置信度至关重要。对于胞内蛋白靶点,活细胞检测需满足两个条件:(1) 药物须穿透细胞膜(通过被动或主动运输);(2) 药物可能与环境中的其他生物分子相互作用。因此,这类检测为测量靶点结合提供了更具生理相关性的系统。细胞裂解液模型虽包含多种蛋白质,但无法反映膜通透性或配体结合的下游效应。本《视角》将涵盖使用离体蛋白、细胞裂解液和活细胞,在苗头化合物鉴定、确证及苗头化合物-先导物优化等早期药物发现阶段量化靶点结合的方法。化学蛋白质组学(第9节)作为快速发展的领域,可在活细胞或裂解液中研究化合物与整个蛋白质组的结合,而不仅限于单一蛋白。
除验证靶点结合外,本文讨论的检测还能测定蛋白-配体结合的热力学、动力学及结构参数。在阐述这些检测技术及其在临床前药物发现中的应用前,我们将首先解析这些参数——它们在指导化合物推进过程中均发挥关键作用。
1.1 药物-靶点热力学
蛋白-药物相互作用可通过单步结合平衡模型描述(图1a)。存在两种表征结合正逆反应的平衡常数:结合常数(KA)与解离常数(KD),二者互为倒数(图1b)。KD单位为摩尔浓度(M),是描述蛋白-配体相互作用最常用的指标——KD值越小(即KA值越大),表明配体与蛋白结合越强,因平衡更倾向于向蛋白-配体复合物方向移动。
KD值测定原理:通过配体对蛋白的滴定实验,确保每个浓度点达到结合平衡(图1c)。图中纵轴表示蛋白结合分数(θ)(最大值为1,代表所有结合位点被占据)。θ=0.5时对应的配体浓度即为KD值,可直接从结合曲线读取。因此,任何能获得与结合程度成正比的响应信号且各浓度点均达平衡的检测方法均可用于KD测定。
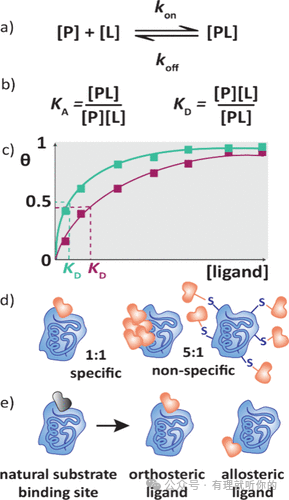
配体对蛋白质的热力学稳定作用可通过量化焓变(ΔH)进行评估。结合过程中的焓变可直接采用等温滴定量热法(ITC)测定,或通过测量解离常数(KD)的温度依赖性并应用范特霍夫分析法推算。蛋白质稳定性的另一关键指标是熔解温度(TM),即蛋白质折叠态与去折叠态呈1:1混合时的温度。多种方法(见第2节)通过检测配体存在下TM的变化来观测蛋白质热稳定性的改变。需特别指出:TM受多种复杂焓/熵因素影响,故不可直接推导KD值。
1.2 药物-靶点动力学
药物-靶点相互作用动力学由药物的结合速率常数(kon)和解离速率常数(koff)决定(图1a)。
·驻留时间(τ)(τ = 1/koff)表示药物与靶点的结合时长,可作为体内药效的预测指标—τ值越大,药物作用持续时间越长。
·共价药物能与靶点不可逆结合,理论上τ趋近无限,其药效实际由靶蛋白的再合成速率决定,从而产生长效治疗作用。
·动力学参数影响选择性:若某药物对同家族多个受体的KD值相同,但对某一靶点的koff更小(即τ更大),则对该靶点的选择性更高
研发启示:
仅依赖热力学参数(如KD)不足以准确预测药物体内行为,需在早期研发阶段测定动力学参数kon和koff以提高成功率。
测定方法:
需通过时间分辨数据(观测信号随时间的变化)计算kon和koff,通常采用表面等离子共振(SPR)等生物传感技术。
1.3结构参数
化学计量比(N)是指蛋白质与配体的比例,即一个蛋白质分子结合多少个药物分子。在大多数情况下,预期比例为1:1,因此通过测定化学计量比可以识别显示非特异性结合的化合物,例如会形成聚集体的化合物以及与多个蛋白质残基结合的混杂性共价化合物(图1d)。
药物分子可以与蛋白质的多种结合口袋相互作用(图1e)。这种结合可以是正构结合,即占据天然底物结合位点;也可以是别构结合,即结合在蛋白质的其他位点上。
确定配体结合位点的金标准是X射线晶体学,尽管冷冻电子显微镜(cryo-EM)在通量和分辨率方面正逐渐成为X射线晶体学的有力竞争者。基于溶液的技术,如蛋白质核磁共振和氢氘交换质谱(HDX-MS)也能提供结合位点信息。
许多检测方法可以采用竞争实验,即在加入药物时观察已知结合口袋中的配体被置换的情况,这为药物与配体结合在相同位点提供了证据。
1.4选择合适的靶点结合检测方法
表1总结了目前可用于确定药物-靶点直接结合的技术,分为以下几类:热分析技术、生物传感技术、质谱技术(MS)、核磁共振技术(NMR)、结构生物学技术、共振能量转移技术(RET)、其他结合检测技术以及化学蛋白质组学技术。同时提供了每种技术的高层次概述,涵盖各检测方法的理论基础,并为表1中的分析和分类提供了参考文献依据。
Table 1.观察蛋白-配体结合的方法
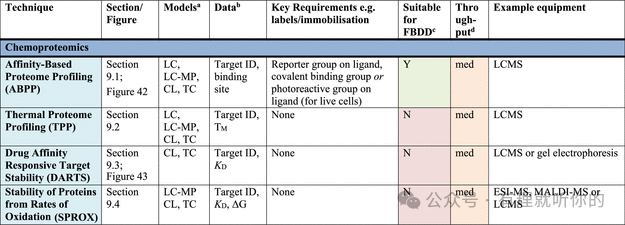
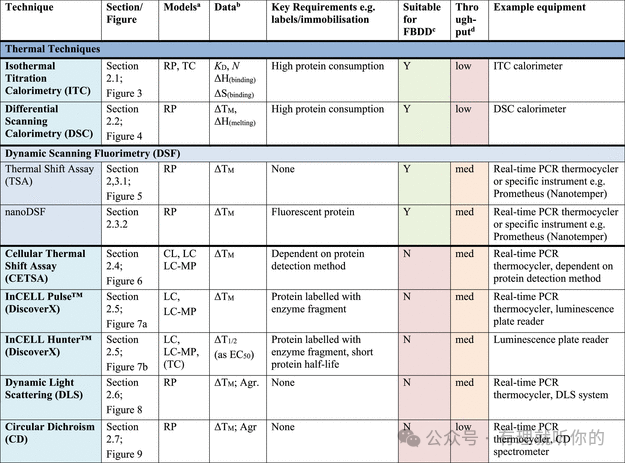
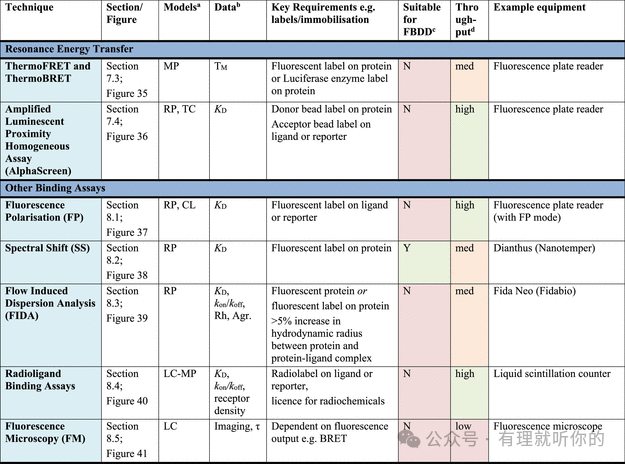
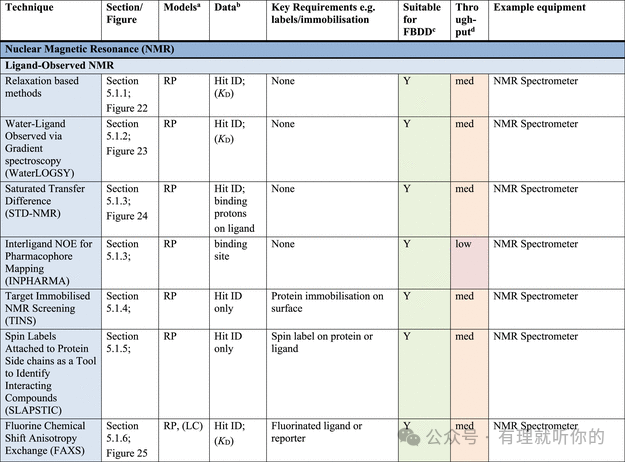
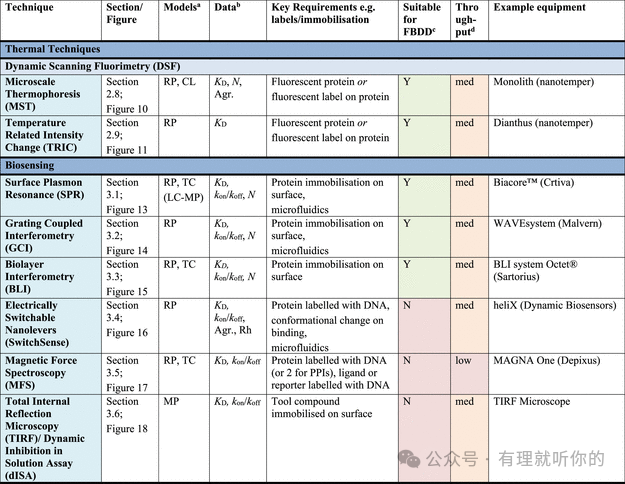
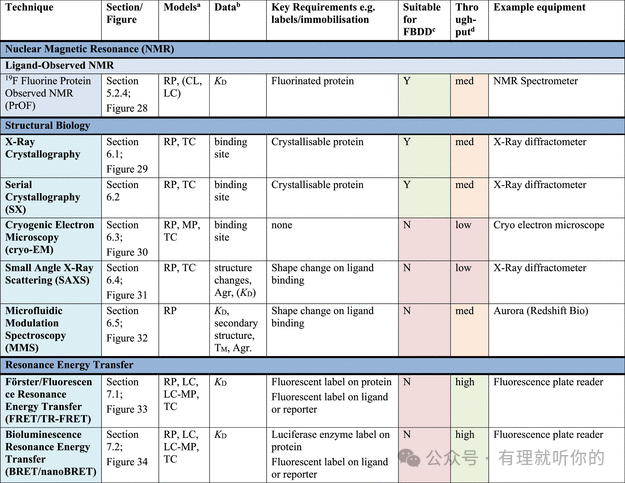
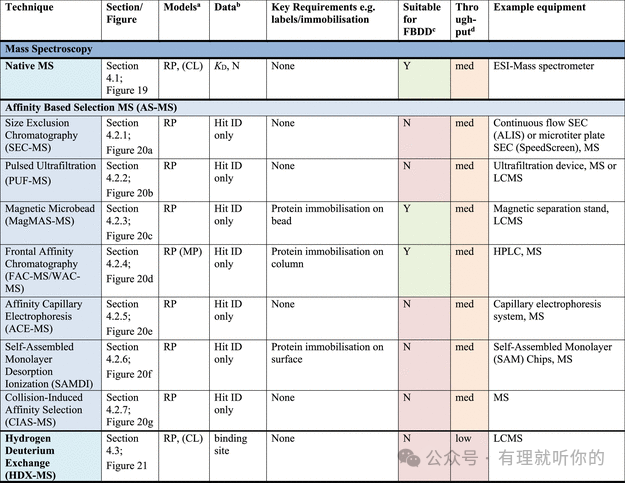
aRP = recombinant protein, MP = membrane protein (solubilized), CL = cell lysate, LC = live cells, LC-MP = live cells-membrane proteins, TC = ternary complex formation (these assays are useful for developing PPI inhibitors, molecular glues and PROTACs). Parentheses indicate some precedence but not standardly performed.
bΔTM = thermal shift, N = stoichiometry, Agr. = aggregation, Rh = hydrodynamic radius, τ = residence time. Parentheses indicate some precedence but not standardly measured.
cSuitability for FBDD is stated when the technique has been commonly used for FBDD, other methods may also be used with further optimization and using high ligand concentrations. Methods suitable for FBDD may also be useful to detect other weak protein–ligand interactions.
dThroughput descriptors are estimates and vary based on instrumentation and protocol but are broadly categorized as follows: high = regularly used for primary screening of large (>50,000) compound libraries, med = suitable for screening of small (1,000–50,000) compound libraries or hit confirmation studies and may be HTS suitable with development, low = currently useful for further compound characterization only.
1.4 靶点结合检测方法的选择
在实验设计方面,通常需要使用工具化合物作为对照。理想的工具化合物应具备稳定性好、溶解性佳(在检测条件下)且能特异性结合靶蛋白特定结合口袋的特性。部分检测方法需要引入标记物/标签,即通过共价修饰连接到蛋白质和/或配体上的报告基团(如荧光染料或特定同位素),以增强检测信号的观察。虽然标记技术可显著提高检测灵敏度,但对蛋白质或配体进行修饰需要专业的技术支持和优化过程。需注意的是,标记物的引入可能会改变蛋白质的结合能力和功能特性,因此"无标记"检测技术具有显著优势。
对于使用标记配体的方法(如荧光偏振FP、生物发光共振能量转移BRET、荧光各向异性筛选FAXS等),通常采用报告分子/探针体系。当竞争性结合配体置换探针时会产生可观测信号,这种方法适用于未标记化合物的筛选。此外,多种检测方法涉及的蛋白质固定化技术也需要专门优化,因为固定化过程可能改变蛋白质构象进而影响配体结合。表1详细列出了各类检测方法的具体要求,包括标记、固定化等特定技术参数。
检测方法的选择具有显著的项目特异性,主要取决于以下因素:
1.靶点特性
2.药物发现阶段(苗头化合物鉴定→苗头化合物确认→苗头到先导→先导化合物优化)
3.专业设备与技术的可获得性
4.试剂与靶蛋白的供应情况
5.项目预算限制
本文后续将基于标准临床前药物研发时间线,系统讨论指导靶点结合检测方法选择的关键因素。
苗头化合物鉴定
药物发现项目可通过多种方法确定起始点,即"苗头化合物"(图2a)。若通过实验方法发现苗头化合物,其筛选策略和靶点结合检测方法的选择主要取决于靶点类别(图2b)。一种苗头化合物鉴定策略是对目标蛋白进行大量分子筛选。20世纪80年代末,实际筛选能力仅为每周数百个化合物。然而,随着技术进步和并行合成方法的采用,筛选能力得到显著提升。到90年代中期,高通量筛选(HTS)已成为制药行业约半数项目的起始点来源。现代HTS化合物库可包含数百万个化合物。为满足这一需求,用于HTS的靶点结合检测方法必须具备适当的微型化和自动化能力。通常采用基于荧光的检测方法(如FRET、BRET、AlphaScreen、FP等),因为这些方法可在酶标仪上实现多通道并行检测。其他靶点结合检测方法也可用于类药分子的化合物筛选,但仅适用于中小规模聚焦库(见表1)。
Figure 2
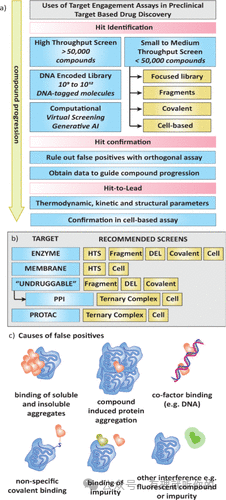
Figure 2. a) Target engagement assays can be used across different stages of the preclinical drug discovery timeline from hit identification with various screening strategies, to confirming hits from a primary screen as well as further characterization of compounds in the hit-to-lead stage. b) Selection of target engagement assay for screening can largely be guided by the class of target. Undruggable targets, which here we also include PPIs, often require different screening strategies than enzymes or membrane-bound proteins. c) False positives in target engagement assays can be caused by many factors.
激酶等酶类蛋白是常见的药物靶点,因其具有明确的结合口袋,可通过生化或生物物理HTS方法轻松鉴定苗头化合物。然而,本文讨论的大多数靶点结合检测方法同样适用于酶类靶点。若不采用高通量筛选,酶类靶点苗头化合物的鉴定方法和检测选择可能取决于其他因素。
靶向GPCR的化合物占FDA批准药物的30%。GPCR等膜结合蛋白的靶点结合研究面临额外挑战,因其在细胞膜外通常不稳定。因此,膜蛋白苗头化合物通常通过基于细胞的检测方法发现,这些方法常通过观察配体结合的下游效应进行功能检测。基于细胞的直接靶点结合检测(如放射性配体结合试验、FRET、BRET、CETSA和InCELL检测)也可用于膜蛋白化合物筛选(见表1"LC-MP"类)。膜蛋白可通过去垢剂溶解于水溶液,针对溶解膜蛋白的特异性检测方法见表1"MP"类(如ThermoFRET/BRET和dISA)。经溶解条件优化后,部分膜蛋白也可用于重组蛋白"RP"类检测。
现代药物发现已逐渐耗尽"可成药"靶点资源,这些靶点通过传统HTS方法取得了巨大成功。因此,研究重点转向曾被视作"不可成药"的靶点。这类靶点通常缺乏明确结合口袋或具有调控蛋白质-蛋白质相互作用(PPIs)的平坦表面,使得基于类药分子的HTS收效甚微。基于片段药物发现(FBDD)提供替代筛选策略:筛选"片段"(分子量通常<300Da)小规模化合物库,作为开发类药分子的起点。片段的小尺寸意味着避免了较大分子中可能阻碍结合的多余功能团,因此更可能发现不可成药靶点的苗头化合物。片段筛选也有助于发现别构位点,这些位点可能比已知底物结合口袋更小更浅。高灵敏度生物物理检测(如NMR、SPR和DSF(TSA))常用于检测片段筛选中的弱结合相互作用。GCI作为新兴技术,对片段筛选和结合动力学测定的灵敏度可能超过SPR。X射线晶体学在FBDD中广泛应用,因为通常需要片段与蛋白结合的原子级结构来指导片段迭代优化为强效类药分子。